
")


Viruses infect organisms across all domains of life, playing key roles in ecological processes such as ocean biogeochemical cycles and the regulation of microbial populations while also causing diseases in humans, animals, and plants. Viruses are Earth’s most abundant biological entities, characterized by rapid evolution, high mutation rates, and frequent genetic exchanges with hosts and other viruses. This constant genetic flux leads to highly diverse genomes with mosaic architectures, challenging functional annotation, evolutionary analysis, and taxonomic classification. Viruses have likely emerged multiple times throughout history despite their diversity, with some lineages predating the last universal common ancestor (LUCA). This highlights a longstanding co-evolutionary relationship between viruses and cellular organisms.
Protein structures, more conserved than sequences, offer a reliable means to study evolutionary relationships and infer gene functions in viruses. However, viral protein structures are significantly underrepresented in public databases, with less than 10% of the Protein Data Bank (PDB) comprising experimental viral protein structures. Recent advances in machine learning, such as AlphaFold2 and ESMFold, have enabled accurate protein structure prediction at scale. Using these tools, researchers have generated a comprehensive dataset of 85,000 predicted structures from 4,400 human and animal viruses, significantly expanding structural coverage. These efforts address the historical gap in viral protein representation, facilitating functional annotation and phylogenetic analysis and shedding light on the evolutionary history of critical viral proteins like class-I fusion glycoproteins.
Researchers from the MRC-University of Glasgow Centre for Virus Research and the University of Tokyo generated 170,000 predicted protein structures from 4,400 animal viruses using ColabFold and ESMFold. They evaluated model quality, performed structural analyses, and explored deep phylogenetic relationships, particularly focusing on class-I membrane fusion glycoproteins, including the origins of coronavirus spike proteins. To support the virology community, they developed Viro3D, an accessible database where users can search, browse, and download viral protein models and explore structural similarities across virus species. This resource aims to advance molecular virology, virus evolution studies, and the design of therapies and vaccines.
The study utilized 6,721 GenBank nucleotide accession numbers, covering 4,407 virus isolates and 3,106 species with host annotations, to extract 71,269 viral protein records. Additional annotations included 4,070 mature peptides, 11,786 protein regions, and 253 polyproteins. Protein structures were predicted using ColabFold and ESMFold, with structural coverage evaluated against the PDB. Proteins were clustered based on sequence and structural similarity, forming 19,067 structural clusters. Functional annotations were expanded using sequence-based and structural networks. A structural similarity map of viral species was created, and comparisons were made with other viral structure databases, highlighting the dataset’s comprehensiveness and structural insights.
The study introduced Viro3D, a robust database encompassing over 170,000 predicted 3D protein structures from 4,400 animal viruses. Using ColabFold and ESMFold, researchers achieved a significant 30-fold increase in structural coverage compared to experimental data. Notably, this dataset revealed functional and evolutionary insights, including the evolutionary origins of coronavirus spike proteins. Structural analyses and protein-protein interaction networks supported functional annotations. Viro3D’s predictions showed high reliability when benchmarked against experimentally solved viral structures. Viro3D provides an unprecedented resource for studying viral evolution, protein function, and structural mechanisms, offering potential applications in antiviral drug and vaccine development.
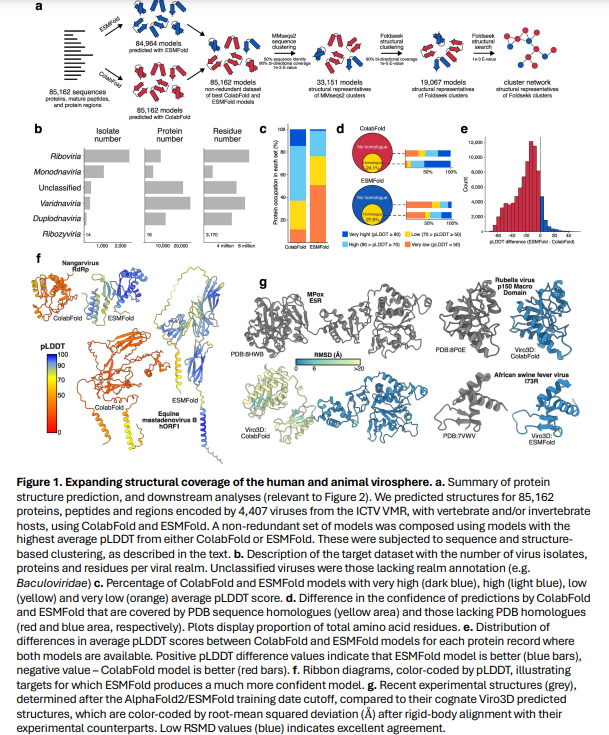
In conclusion, the study expanded viral protein structural coverage 30-fold by modeling 85,000 proteins from 4,400 human and animal viruses, with 64% of models being highly confident. Combining ColabFold and ESMFold methods enhanced efficiency, accuracy, and speed. Structural clustering reduced viral diversity to 19,000 distinct structures, 65% unique to this dataset, with many found near viral genome ends, suggesting evolutionary hotspots. Analysis revealed that viral proteins often lack homologs in cellular organisms, indicating extensive remodeling. The study traced their evolution by exploring class-I fusion glycoproteins, highlighting their role in virus transmission and pathogenesis, and offering valuable insights for virology research.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. Don’t Forget to join our 60k+ ML SubReddit.
Sana Hassan, a consulting intern at Marktechpost and dual-degree student at IIT Madras, is passionate about applying technology and AI to address real-world challenges. With a keen interest in solving practical problems, he brings a fresh perspective to the intersection of AI and real-life solutions.



")






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leave a comment